|
Myotonic Dystrophy is a slowly progressive neuromuscular Disease and the symptoms of the disease do gradually get worse over time. However, there are a number of ways to manage the symptoms of the disease and more and more management techniques are becoming available. In the very long-term outlook most people with DM will have shorter life expectancies, One study available at the end of this post gives an average of 59 years for females with DM1 and 60 Years for males
For children with the Congenital form once beyond the early problems of respiratory distress the prognosis for life is relatively good. There will be improvement in early childhood and children will make steady progress in these early years. Motor function will improve, most children will walk and the marked hypotonia or floppiness will improve or disappear. For most children their performance will be limited by mental capacity and not by physical handicap.
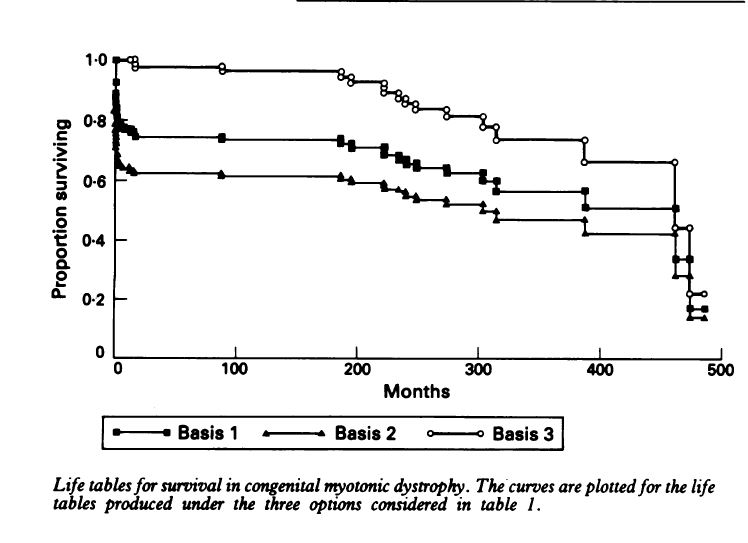
Reardon reports on the outcomes of 115 patients with confirmed diagnosis’s of CMD. The data suggests a 25% of death before 18 months of age and a 50% chance of survival into the mid-30’s. tables that follow are from Reardon. The Natural History of Congenital myotonic Dystrophy: Mortality and long term aspects. Archives of Disease in Childhood 1993; 68:177-181
Long-term Survival Probability
|
Age X
|
Probability of Survival to Age X
|
Age X
|
Probability of Survival to Age X
|
|
0
|
1.000
|
|
|
|
Days
|
|
|
|
|
1
|
.922
|
|
|
|
2
|
.887
|
|
|
|
3
|
.878
|
|
|
|
7
|
.861
|
|
|
|
10
|
.861
|
|
|
|
12
|
.843
|
|
|
|
20
|
.835
|
|
|
|
21
|
.826
|
|
|
|
Months
|
|
|
|
|
1
|
.807
|
234 |
.671 |
|
2
|
.791
|
240 |
.657 |
|
3
|
.791
|
249 |
.643 |
|
4
|
0774
|
274 |
.625 |
|
7
|
.765
|
304 |
.598 |
|
12
|
.765
|
315 |
.564 |
|
14
|
.757
|
388 |
.508 |
|
16
|
.745
|
462 |
.339 |
|
88
|
.737
|
474 |
.169 |
|
186
|
.724
|
486 (40 yr.) |
0 |
|
195
|
.711
|
|
|
|
222
|
.698
|
|
|
|
223
|
.685
|
|
|
There was no instance of a congenitally affected patient having children. 48 patients reached the age of 20, 12 reached the age of 30. (of 115 patients). However, Reardon noted that “The degree of precision of estimated survival probabilities at the far end of the life table is recognized to be extremely poor” (or to put is more clearly he’s not sure how good the estimates are in the older years 30-40’s)During the study only 3 such individuals were studied and all died in this age band.
Details of Current Employment among the 48 patients reaching 20 years of Age
| Employment |
No of Patients |
| Local Government Clerical Worker |
1 |
| Cinema Attendant |
1 |
| Garden Center Operative |
1 |
| Adult Training Workshop |
5 |
| Housewife |
2 |
| Unemployed |
38 |
Most of the patients with CMD will be unemployed.
Chronic Medical Problems in surveying Patients n=71
| Problem |
Number of Patients |
Percentage |
| Constipation/Diarrhea |
24 |
34% |
| Recurrent Otitis |
15 |
21% |
| Scoliosis |
7 |
10% |
| Cardiac Rhythm Disturbance |
3 |
4% |
| Vesicoureteric Reflux |
1 |
1% |
Causes of Death n=44
| Cause |
No of Deaths |
| Constipation/Diarrhea |
34% |
| Recurrent Otitis |
21% |
| Scoliosis |
10% |
| Cardiac Rhythm Disturbance |
4% |
| Vesicoureteric Reflux |
1% |
However, during late childhood and early adolescence the “adult” features of myotonic dystrophy appear. Eye problems can be early on detected by the second decade and progressively muscles will be detected gradually weakening. No case of CMD has been found not to develop the adult version. O’Brien reports that of 46 patients with CMD 4 died outside the neonatal period at 4, 18, 19 and 22 years. Four more were considered seriously disabled with a poor prognosis. Problems seen in this group of 30 were gastrointestinal Problems in 8 (Constipation and abdominal pain) Talipes in 5. None in the group had any children and males showed marked testicular tubular failure indicating significant reproductive problems. Thus, long-term prognosis from a medical viewpoint is not bright.
Reardon noted that because of gastrointestinal problems children with CMD will show a positive anal dilation test. This test is sometimes used to diagnosis sexual abuse. Parents with CMD should be aware of this information. This test should not be used for any conclusive evidence of sexual abuse with children with CMD.
However, children with CMD are generally happy and cheerful individuals with a distinct personality and add a lot to family life as a whole. Medical outlook should be integrated with other family factors (see personal stories)
New Study May 1999:
A 10-year study of mortality in a cohort of patients with myotonic dystrophy.
Mathieu J; Allard P; Potvin L; Pr´evost C; B´egin P
Neuromuscular Clinic, Complexe Hospitalier de la Sagamie, Quebec University in Chicoutimi, Canada.
Neurology, 52(8):1658-62 1999 May 12
Abstract
OBJECTIVE: To determine the age and causes of death as well as the predictors of survival in patients with myotonic dystrophy (DM). METHODS: In a longitudinal study, a cohort of 367 patients with definite DM was followed for 10 years. RESULTS: During the 10-year period, 75 of the 367 DM patients (20%) died. The mean age at death (53.2 years, range 24 to 81) was similar for men and women. Among these 75 patients, 32 (43%) died of a respiratory problem, 15 (20%) of cardiovascular disease, 8 (11%) of a neoplasia, and 8 (11%) died suddenly. The ratio of observed to expected deaths was significantly increased to 56.6 (95% confidence interval [CI] 38.7 to 78.0) for respiratory diseases, 4.9 (95% CI 2.7 to 7.7) for cardiovascular diseases, and 2.5 (95% CI 1.1 to 4.6) for neoplasms. The mean age at death was 44.7 years for the childhood phenotype of DM, 47.8 years for the early-adult, 55.4 years for the adult, and 63.5 years for the mild phenotype (F = 4.8, p = 0.005). The age-adjusted risk of dying was 3.9 (95% CI 1.3 to 11.0) times greater for a patient with a distal weakness and 5.6 (95% CI 2.2 to 14.4) times greater for a patient with proximal weakness as compared with a person without limb weakness. CONCLUSIONS: Life expectancy is greatly reduced in DM patients, particularly in those with early onset of the disease and proximal muscular involvement. The high mortality reflects an increase in death rates from respiratory diseases, cardiovascular diseases, neoplasms, and sudden deaths presumably from cardiac arrhythmias.
Full text of Article above
Here is another study abstract on Longevity:
Age and causes of death in adult-onset myotonic dystrophy.
Author
de Die-Smulders CE; H¨oweler CJ; Thijs C; Mirandolle JF; Anten HB; Smeets HJ; Chandler KE; Geraedts JP
Address
Department of Clinical Genetics, Academic Hospital Maastricht, The Netherlands. christine.dedie@gen.unimaas.nl
Source
Brain, 121 ( Pt 8)():1557-63 1998 Aug
Abstract
Myotonic dystrophy is a relatively common type of muscular dystrophy, associated with a variety of systemic complications. Long term follow-up is difficult because of the slow progression. The objective of this study was to determine survival, age at death and causes of death in patients with the adult-onset type of myotonic dystrophy. A register of myotonic dystrophy patients was set up in Southern Limburg (the Netherlands), using data longitudinally collected over a 47-year period (1950-97). Survival for 180 patients (from the register) with adult-onset type myotonic dystrophy was established by the Kaplan-Meier method. The median survival was 60 years for males and 59 years for females. Survival of the patients was also estimated from the age of 15 years to the ages of 25, 45 and 65 years and compared with the expected survival of age- and sex-matched birth cohorts from the normal Dutch population. The observed survival to the ages of 25, 45 and 65 years was 99%, 88% and 18% compared with an expected survival of 99%, 95% and 78%, respectively. Thus, survival to the age of 65 in patients with adult-onset myotonic dystrophy is markedly reduced. A weak positive correlation between the CTG repeat length and younger age at death was found in the 13 patients studied (r = 0.50, P = 0.08). The cause of death could be determined in 70 of the 83 deceased patients. Pneumonia and cardiac arrhythmias were the most frequent primary causes of death, each occurring in approximately 30%, which was far more than expected for the general Dutch population. In addition, we assessed mobility in the years before death in a subgroup of 18 patients, as a reflection of the long-term physical handicap in myotonic dystrophy patients. Half of the patients studied were either partially or totally wheelchair-bound shortly before their death.
|