A new study of myotonic dystrophy patients indicates that an older style test of maximum voluntary ventilation may help Myotonic Dystrophy patients know their lung function better.
The maximal voluntary ventilation (MVV) is another measure of the neuromuscular and respiratory systems. The MVV is the total volume of air exhaled during 12 sec of rapid, deep breathing, which can be compared with a predicted MVV defined as the forced expiratory volume in 1 sec (FEV1) × 35 or 40. A significant difference between the predicted and measured MVV may indicate insufficient neuromuscular reserve, abnormal respiratory mechanics, or an inadequate effort. Progressive reduction of tidal volumes during the test is consistent with neuromuscular abnormalities but also occurs with gas trapping as a result of disorders that cause airflow limitation.
The MDF recruited 15 international clinicians with long-standing experience in the care of DM2 patients to develop consensus-based care recommendations. The single text procedure was adopted. This process generated a 4-page Quick Reference Guide and a comprehensive 55-page document that provides care recommendations for DM2 patients.
Summary The resulting recommendations will help standardize and improve care for DM2 patients and facilitate appropriate management in centers without neuromuscular specialists.
There is a new compound that is being tested to see if it can help with congenital myotonic dystrophy. It is being tested for a number of applications including tooth repair and Alzheimer’s and just might help with the congenital form of myotonic dsytrophy. this is a molecule being developed by AMO pharma.
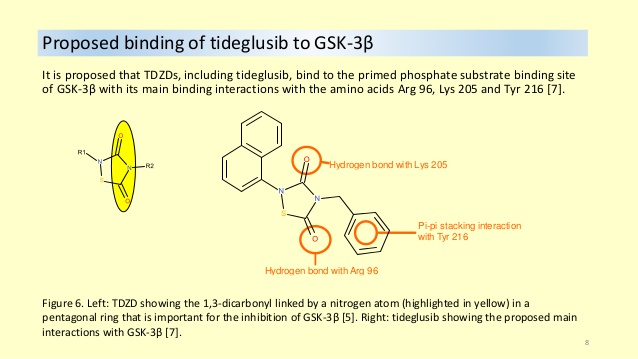
Tideglusib (NP-12, NP031112) is a potent, selective and irreversible[1] small molecule non-ATP-competitive glycogen synthase kinase 3 (GSK-3) inhibitor.
Tooth repair mechanisms that promotes dentine reinforcement of a sponge structure until the sponge biodegrades, leaving a solid dentine structure. In 2016, the results of animal studies were reported in which 0.14 mm holes in mouse teeth were permanently filled.[7]
Tideglusib is being studied in Phase II clinical trials as a treatment for congenital/juvenile-onset myotonic muscular dystrophy type I.[8]
There is a clinical study that will be starting shortly and you might be able to participate when this trial opens. Click here for more information.
This is a fairly scientific study of using the CRISPER Technology to help find a treatment for myotonic dystrophy. DM1 is caused by expanded CTG repeats in the 30 UTR. It is conceivable that simply deleting the expanded CTG repeats may cure the disease. With the advancement of therapeutic genome-editing technologies, this is becoming more realistic.
IMAGE: SCRIPPS RESEARCH GRADUATE STUDENT ANGEL ANGELBELLO AND CHEMISTRY PROFESSOR MATTHEW D. DISNEY, PHD, REPORT PROMISING DATA ABOUT THEIR RNA-BINDING MYOTONIC DYSTROPHY TYPE 1 COMPOUND IN PNAS. view more
CREDIT: SCRIPPS RESEARCH
JUPITER, Fla. – April 1, 2019 — People diagnosed with myotonic dystrophy type 1 have difficulty unclenching muscles due to a genetic defect that generates toxic material within their cells. There is currently no treatment. In a new report published in the Proceedings of the National Academy of Sciences on Monday, a group at Scripps Research in Florida reports making a potential drug that targets its key disease-causing RNA. In mouse and cellular models of myotonic dystrophy type 1, it improved the muscle defects with no apparent side-effects.
The study appeared on line Friday in PNAS. More work lies ahead before testing in people will be possible, but “the results look better than we could have imagined,” says lead author Matthew Disney, PhD, a Scripps Research chemistry professor.
“The results suggest that our technology can be used to treat myotonic dystrophy type 1 and similar categories of inherited diseases, and without unintended, off-target effects,” Disney says, adding, “There is great potential for drugging RNA in all disease settings.”
The most common form of muscular dystrophy in adults, myotonic dystrophy type 1 has been estimated to affect around 1 in 8,000 people, although the Myotonic Dystrophy Foundation, based in San Francisco, reports that misdiagnosis is common, likely leading to underreporting. Genetic studies suggest the actual numbers are more than three times higher, around 1 in 2,500, says Molly White, CEO of the foundation.
The disease is inherited. Symptoms emerge in late teens or early adulthood as genetic changes accumulate. They include muscle clenching, lock-jaw, early-onset cataracts, brain fog, muscle wasting and weakness, digestive difficulties, and sudden cardiac death, White says. Severity and rate of progression depend on factors including the nature of the genetic defect.
Myotonic dystrophy type 1 occurs when a sequence of three nucleotides, CTG, is repeated too many times within a gene called DMPK. Toxic protein clumps generate further genetic defects, resulting in muscle weakness and other symptoms. A healthy person could carry between 5 and 35 repeats of CTG within that gene without experiencing obvious difficulty. But symptomatic people may have 50, 100 or even up to 4,000 repeats of the CTG sequence.
The drug Disney’s group designed, called Cugamycin, works by recognizing the toxic RNA repeats and destroying the garbled gene transcript. Significantly, in treated animals, the drug left the healthy version of the gene transcript intact. The results were consistent in both the mouse model of myotonic dystrophy type 1 and in human patient-derived muscle fibers called myotubes.
Cugamycin was made by attaching an RNA-binding molecule to an existing drug called bleomycin, which cleaves nucleic acids.
“Analysis of the tissue from a pre-clinical disease model showed more than 98 percent of the disease defects are improved, with no detectable off-targets.” Disney says.
So far, the results have been excellent, but these studies are still in their early stages, says Alicia Angelbello, the study’s first author and a Scripps Research graduate student.
“A key next question will be to evaluate the effectiveness of our compound over a longer period of time,” she adds. In the current study, the treated mice experienced fewer instances of “myotonic discharges” in their muscles–occasions when it takes longer than usual for a contracted muscle to relax–compared to untreated mice, Angelbello says.
“Once given the Cugamycin at a dose of 10 milligrams per kilogram, the frequency of the myotonic discharges was reduced by 50 percent, which is a significant improvement,” Angelbello says. “The fact that we can improve the muscular and genetic defects in DM1 mice with the molecules we have made in the lab is a significant step in learning about how to treat this disease,” she says. The Myotonic Dystrophy Foundation has supported Disney’s work for many years.
“Matt Disney is super-committed to making a treatment for this disease,” says White, CEO of the Myotonic Dystrophy Foundation. “It’s clear that many people think his lab is on the right track. We’re thrilled at the progress he is making.”
###
In addition to Disney and Angelbello, the authors of the study, “Precise small molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model,” include Suzanne Rzuczek, Jonathan Chen and Michael Cameron of Scripps Research; Kendra McKee, Hailey Olafson and Eric Wang of the University of Florida; and Walter Moss of Iowa State University. Disney and Wang consult for Expansion Therapeutics. Rzuczek is an employee of Expansion Therapeutics. Disney’s work was funded by the National Institutes of Health (grant DP1NS096898) and the Muscular Dystrophy Association (grant 380467). The Myotonic Dystrophy Foundation also provided support.